人工智能驱动的方法挑战蛋白质结构的传统观点
在《自然通讯》最近发表的一篇文章中,一个团队提出了一种人工智能驱动的方法来探索蛋白质领域的结构相似性和关系。该团队的成员来自弗吉尼亚大学,包括数据科学学院院长 Phil Bourne、学院高级科学家 Cam Mura 和 UVA 近期校友 Eli Draizen。
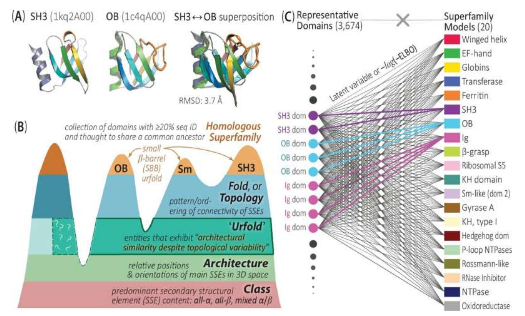
他们的研究挑战了关于蛋白质结构关系(即相似性和差异性模式)的传统观念,并在此过程中发现了许多传统方法所忽略的微弱关系。
具体来说,作者报告了一个计算框架,该框架可以以新颖、灵活和细致入微的方式大规模检测和量化这种蛋白质关系(跨越无数蛋白质),该框架将基于深度学习的方法与一种称为 Urfold 的新概念模型相结合,允许两种蛋白质尽管具有不同的拓扑结构或“折叠”,但仍表现出结构相似性。
Bourne、Mura 和 Draizen 与 Stella Veretnik 合作完成了该项目。所有作者都是 Bourne & Mura 计算生物科学实验室的成员,该实验室隶属于数据科学学院和 UVA 生物医学工程系。
该出版物是 Bourne 实验室多年工作的成果,旨在开发名为 DeepUrfold 的人工智能驱动框架,以便系统地、大规模地探索 Urfold 结构关系理论。
利用 DeepUrfold,Bourne 实验室团队检测到了蛋白质世界中那些原本被视为不相关的蛋白质之间的微弱结构关系,无论是在进化上还是在其他方面。
在捕捉和描述这些远距离关系时,DeepUrfold 从“社区”的角度看待蛋白质关系,避免了将蛋白质分类到单独的、不重叠的箱体中的传统方法。总而言之,这些新的方法可以推动研究人员超越以静态、几何术语思考蛋白质相似性,转向更综合的方法。
免责声明:本答案或内容为用户上传,不代表本网观点。其原创性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容、文字的真实性、完整性、及时性本站不作任何保证或承诺,请读者仅作参考,并请自行核实相关内容。 如遇侵权请及时联系本站删除。